氨基化石墨烯改性水性聚氨酯的合成及性能
胡 令 1 ,蒋平平 1* ,张萍波 1 ,卞 刚 1 ,盛松松 1 ,黄 敏 1 ,夏嘉良 2
( 1. 江南大学 化学与材料工程学院 食品胶体与生物技术教育部重点实验室,江苏 无锡 214122; 2. 昆山嘉力普制版胶粘剂油 墨有限公司,江苏 昆山 215300)
摘要: 用 3-氨丙基三甲氧基硅氧烷( APTMS) 插层改性氧化石墨烯( GO) ,得到氨基化石墨烯( APTMS-GO) 。通过 FTIR、XRD、Raman、TG、TEM、XPS 表征了 APTMS-GO 的结构和形态。将 APTMS-GO 与带有异氰酸根的聚氨酯预 聚物以原位聚合的方式聚合,制得了含 APTMS-GO 质量分数( 以聚氨酯合成原料的总质量计,下同) 为 0、 0. 06%、0. 11%、0. 16%、0. 22%、0. 33%和 0. 55%的 APTMS-GO/WPU 纳米复合材料,并测试了其拉伸性能、热性 能和疏水性的变化; 利用 FESEM 和 TEM 观察了截面中纳米填充物的分散情况及乳液的粒径。结果表明: 通过 原位聚合得到的复合材料拉伸强度明显改善,由纯水性聚氨酯的10. 13 MPa 增加到28. 96 MPa; 当 APTMS-GO 质 量分数为 0. 22%时,复合材料的初始分解温度( T d5 ) 增大到 279 ℃,与纯水性聚氨酯相比提高了 34 ℃; 随着 APTMS-GO 质量分数的逐步增大,复合膜的接触角由 71. 3°提高到 91. 28°,复合材料的疏水作用得到了改善。
关键词: 水性聚氨酯; 氨基化石墨烯; 原位聚合; 复合材料; 疏水性; 功能材料
中图分类号: TQ323. 8 文献标识码: A 文章编号:1003 -5214( 2017) 01 -0020 -09
水性聚氨酯( WPU) 因以水为介质,具有无毒、 无污染、价廉等优点,已被广泛应用于涂料、粘合剂、 塑料、橡胶、纤维和皮革涂饰剂等领域 [1] 。水性 WPU 制备过程中亲水性基团的引入导致制备的涂 层易吸潮,疏水性差,热学、力学等性能欠缺。因此, 需对其改性处理,WPU 常见改性方法分为有机改性 与无机改性。目前,国内外对于有机改性主要集中 在环氧树脂改性 [2] 、丙烯酸酯改性 [3] 、有机硅改 性 [4] 以及有机氟改性 [5] ,而无机改性主要集中在无 机纳米粒子改性 [6 -13] 。纳米粒子具有尺寸小、比表 面积大的优点,能够产生量子效应和表面效应,使制 得的纳米复合材料较常规复合材料具有更优异的物 理性能与力学性能。
石墨烯具有高弹性、高模量、高导热率及高导电 率等优点 [14 -18] ,其在高分子领域中作为填料得到了 广泛的应用,但石墨烯易团聚,与基体的相容性差, 导致石墨烯的应用受到了一定限制。共价键改性和 非共价键改性是促进石墨烯在高分子中应用的重要 手段。作为石墨烯衍生物的氧化石墨( GO) ,表面的 含氧基团使其易被修饰且能够很好地分散于水中, 从而能应用到水性聚氨酯的改性中。
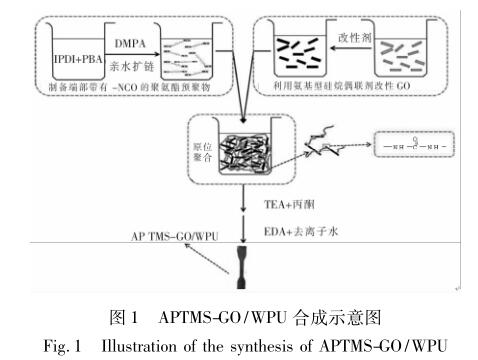
本文利用 3-氨丙基三甲氧基硅氧烷( APTMS) 与 GO 表面的环氧键发生亲核加成反应,使 GO 表 面嫁接上氨基,将得到的带有氨基的 APTMS-GO 以 不同的浓度与带有异氰酸根的聚氨酯预聚物通过原 位聚合的方式聚合,得到相间有效接触且共价连接 的有机 - 无机纳米复合物,在一定程度上改善了聚 氨酯的热性能、力学性能和疏水性能。
1 实验部分
1. 1 原料、试剂与仪器
鳞片石墨( 平均粒径44 μm) ,青岛金日来石墨有 限公司; 异氟尔酮二异氰酸酯( IPDI,质量分数为 97%) ,工业纯,无锡东润电子材料科技有限公司; 聚己 二酸丁二酯2000( PBA -2000) ,青岛宇田化工有限公 司;2,2-双( 羟甲基) 丙酸( DMPA) ,质量分数为98%,阿 拉丁试剂( 上海) 有限公司;3-氨丙基三甲氧基硅氧烷 ( APTMS) ,AR,百灵威科技有限公司; 浓硫酸( 质量分 数为98%) 、盐酸、辛酸亚锡、无水乙二胺( EDA) 、N-甲 基吡咯烷酮( NMP) 、三乙胺( TEA) 、丙酮,AR,国药集团 化学试剂有限公司; 去离子水,自制。
Nicolet 670 型全反射傅里叶红外光谱仪,美国 Thermo 公司; TGA/DSC1/1100SF 型热重分析仪,瑞 士 Mettler - Toledo 公司; WDT -10 型微机控制电子 万能试验机,深圳凯强力机械有限公司; D8 型 X 射 线多晶衍射分析仪( XRD) ,德国 Bruker AXS 有限公 司; Multimode 8 型 原 子 力 显 微 镜 ( AFM) ,德 国 Bruker 公司; S -4800 型场发射扫描电镜( FESEM) , 日本日立株式会社; inVia 型显微共聚焦拉曼光谱仪 ( Raman) ,英国 Renishaw 贸易有限公司; Escalab 250 型电 子 能 谱 分 析 仪 ( XPS) ,美 国 Thermo - VG Scientific 公司; Zeta PALS 型 Zeta 电位/纳米粒度分 析仪,美国 Brookhaven 公司; JEM - 2100 型透射电 子显微镜( TEM) ,日本电子株式会社; 5565 型电子 万能材料试验机,美国 Instron 公司。
1. 2 氧化石墨烯( GO) 的制备
采用改进的 Hummers 法制备 GO [19] : 将 2. 5 g 片状石墨和 1. 88 g NaNO 3 加入到 1 L 三口烧瓶中, 在冰水浴中边搅拌边缓慢滴加 187 mL 浓 H 2 SO 4 ,再 缓慢加入11. 3 g KMnO 4 ( 1 h 内加完) ,继续搅拌2 h 后,在室温下继续搅拌5 d。边搅拌边缓慢滴加质量 分数为 5%的稀硫酸 350 mL( 滴加时间超过 1 h) , 接着在 98 ℃下继续搅拌 2 h,待系统降温到 60 ℃, 再滴加质量分数为30%的双氧水7. 5 mL,继续搅拌 2 h。混合液离心分离,先用 V( H 2 SO 4 ) ∶V( H 2 O 2 ) = 2∶1 的 500 mL 3% H 2 SO 4 -0. 5% H 2 O 2 ( 均为质量分 数) 混合液洗涤,重复离心、洗涤 15 次; 再用 500 mL 质量分数为3% 的 HCl 洗涤3 次,用500 mL 去离子 水洗涤 1 次,将得到的产物超声 30 min 溶解在 500 mL 水中,水溶液用弱碱性离子交换树脂除去残余的 HCl,经冷冻干燥得到 1. 8 g GO。
1. 3 氨基化石墨烯( APTMS-GO) 的制备
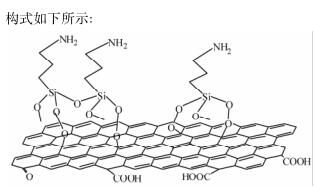
参照文献[20]制备 APTMS-GO: 将 1 g GO 分散 到 300 mL 乙醇中,超声 0. 5 h,随后将其加入到 500 mL 的单口烧瓶中,然后缓慢滴加 1. 79 g( 10 mmol) APTMS,混合物在 800 r/min 下加热回流 6 h,之后 冷却到室温,去掉上层清液,底物用乙醇和水分别洗 涤 3 次,最后冷冻干燥得到 1. 71g APTMS-GO,其结构式如下所示:
1. 4 APTMS-GO/WPU 纳米复合材料的制备
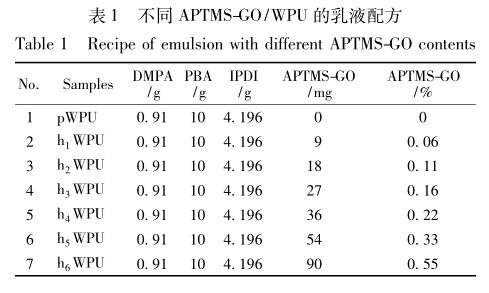
将 9 mg APTMS-GO〔占聚氨酯合成原料( 溶剂 除外) 质量的 0. 06%〕分散到 10 mL NMP 中,室温 下超声 3 h; 同时,在装有搅拌器、冷凝管、氮气导入 装置的250 mL 圆底烧瓶中,加入经真空脱水的10 g ( 0. 005 mol) PBA,升温至 80 ℃,待 PBA 全部溶解 后,加入2. 7 mL( 4. 196 g,0. 02 mol) IPDI,80 ℃下反 应 1 h,加入 0. 91 g( 0. 007 mol) DMPA 进行扩链,80 ℃下反应3 h,得到带有异氰酸根的预聚物; 然后,滴 加 5 mg 辛酸亚锡,并将之前已分散好的 APTMS-GO 加入到该混合液中进行原位聚合,剧烈搅拌1 h。降 温至50 ℃,加入0. 688 g( 0. 007 mol) TEA( DMPA 的 中和度达 100%) ,同时添加 10 mL 丙酮调节黏度, 45 min 后,降温至 10 ℃以下,随之向其中加入溶有 0. 425 g( 0. 007 mol) EDA 的 42 mL 去离子水,在 1 800 r/min 高速搅拌下进行低温乳化 1 h,得到 APTMS-GO 质量分数为 0. 06% 的 APTMS-GO/WPU 乳液。多次进行以上操作,只改变 APTMS-GO 的用 量,合 成 了 pWPU、0. 06% APTMS-GO/WPU ( h 1 WPU ) 、0. 11% APTMS-GO/WPU ( h 2 WPU ) 、 0. 16% APTMS-GO/WPU ( h 3 WPU) 、0. 22% APTMS- GO/WPU ( h 4 WPU ) 、 0. 33% APTMS-GO/WPU ( h 5 WPU) 、0. 55% APTMS-GO/WPU( h 6 WPU) 7 组 乳液,其中 APTMS-GO 的添加量均为质量分数,7 组 乳液相应的配方见表 1,合成操作示意图见图 1。将 已制备好的乳液均匀地注入到聚四氟乙烯板上,放 入烘箱中,50 ℃恒温干燥 48 h,即可得到乳胶膜。
1. 5 测试与表征
红外光谱: 扫描范围为4 000 ~ 500 cm -1 ; 热重 测试: 氮气为载气,流量 50 mL/min,测试温度 50 ~ 600 ℃,升温速率20 ℃ /min,样品质量为5 ~10 mg; XRD: Cu 靶 K α ,功率 2. 2 kW,扫描范围 3° ~50°,扫 描速度 2( °) /min; 原子力显微镜( AFM) : 云母片为 基底,Tapping 模式; 显微共聚焦拉曼光谱: 扫描范围 100 ~4 000 cm -1 ,激发波长 532 nm; X 射线光电子 能谱分析( XPS) : 辐射源为 Al K α ( hν =14 846 eV) , 分析室真空度为 13 310Pa; 拉伸测试: 拉伸速率 50 mm/min,每个样测3 次,取平均值; TEM: 测试电压3 kV,铜网为基底; 场发射扫描电子显微镜( FESEM) : 测试电压 2 kV,测试前样品需在液氮中淬断并进行 表面喷金处理; 疏水性测试: 接触角范围 0° ~180°, 精度 ±0. 1 ℃,室温。
2 结果与讨论
2. 1 APTMS-GO 的表征2. 1. 1 FTIR 分析
GO 与 APTMS-GO 的红外光谱如图 2 所示。
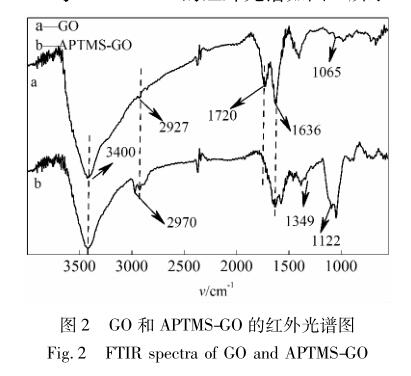
图 2a 中出现的3 400、2 927、1 720、1 636、1 065 cm -1 处的特征峰 [19] ,分别对应于 GO 上—OH 的伸缩振动、—CH 2 的伸缩振动、GO 片层周围的羧基或 羰基的 C O 伸缩振动以及— C C—的骨架振动 和 C—O 的伸缩振动,以上官能团的出现说明鳞片 石墨被成功地氧化。而在图2b 中1 349、1 122 cm -1 处出现的特征峰 [20] 则分别归属于 C—N 的伸缩振 动和 Si—O—C 的伸缩振动,其中,1 349 cm -1 峰强 度较弱,分析可能是由于硅烷偶联剂的 C—N 键属 于峰强较弱的脂肪胺,且 APTMS 与 GO 表面的环氧 亲核加成后,影响了其峰强度。3 400、1 720 cm -1 处 的峰强度也明显减弱。以上新峰的出现和原有峰强 度的变化都说明 APTMS 影响了 GO 的化学结构。
2. 1. 2 XRD 分析
图 3 为 GO 与 APTMS-GO 的 XRD 谱图。
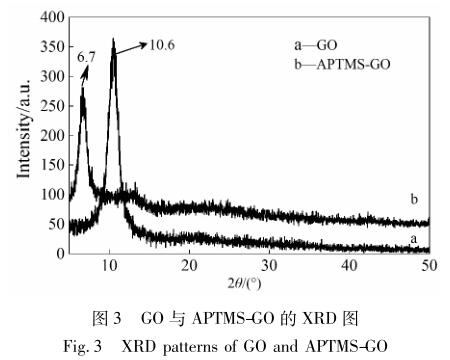
从图 3 可以看出,2θ = 10. 6°出现尖锐的特征 峰,对应于氧化石墨烯的( 001) 晶面,而通过 APTMS 改性后,该峰位出现了转移,从 10. 6° 转移到了 6. 7°,相应的层间距也发生了改变,根据布拉格方程 计算得出,层间距由原来的 0. 83 nm 扩大到了 1. 31 nm,由此可知,硅烷偶联剂被成功地插层到了氧化 石墨烯片层当中 [21] 。
2. 1. 3 Raman 分析
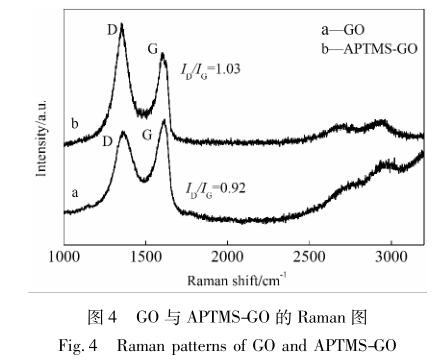
拉曼光谱是用来表征石墨烯片层上有序无序度 的一种表征技术,图 4 为 GO 和 APTMS-GO 的拉曼 光谱图。
图 4 中,GO 的 D 峰和 G 峰分别位于1 356、 1 598 cm -1 处,2D 峰与 G + D 峰,分别位于2 703、 2 948 cm -1 处。其中,D 峰是由 C—C 的无序振动引 起的,表征 sp 3 杂化结构的碳原子; G 峰是由 C—C 的伸缩振动引起的,表征石墨 sp 2 结构的碳原子,2D 峰与 D + G 峰的出现则说明了所制备的 GO 层数较 少。I D /I G 值则表示石墨烯面内无序结构和有序结 构的比值。由图 4 可知,经过 APTMS 处理后,I D /I G 值由 GO 的0. 92 增大到 APTMS-GO 的1. 03,说明氧 化石墨烯原有 sp 2 杂化碳原子部分转变成了 sp 3 杂 化碳原子,反映出原有层面的破坏。由此可知, APTMS 在反应过程中破 坏 了 氧 化 石 墨 烯 的 表 面 [21] 。
2. 1. 4 热重分析
图 5 为 GO 与 APTMS-GO 的热失重曲线。
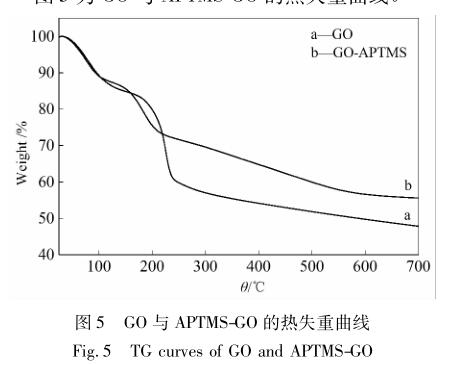
由图5 可知,在100 ℃之前,两条曲线均出现了 热损失,可能是石墨烯片层上吸附的水分子脱附造 成的。在 200 ℃左右,GO 出现了最大的热失重,这 对应于氧化石墨烯片层上含氧官能团的热分解。相 比 GO 在 700 ℃处 52. 18%的热损失,APTMS-GO 则 只有 44. 42%的热损失。APTMS-GO 热稳定性的提 高归 因 于 APTMS 成 功 嫁 接 到 GO 上,产 生 的 Si—O—Si键或者 Si—O—C 键将其热稳定性赋予给 了整个石墨烯片层,最后提高了其热稳定性 [21] 。 APTMS-GO 在 700 ℃ 的 残 余 量 为 55. 58%,而 APTMS 结构中 Si—O—Si 的分子质量占 APTMS 分 子质量的 67. 65%。可以推断出,APTMS 嫁接到 GO 上的质量百分比为 82. 16% [22] 。
2. 1. 5 AFM 尺寸与 TEM 形貌观察
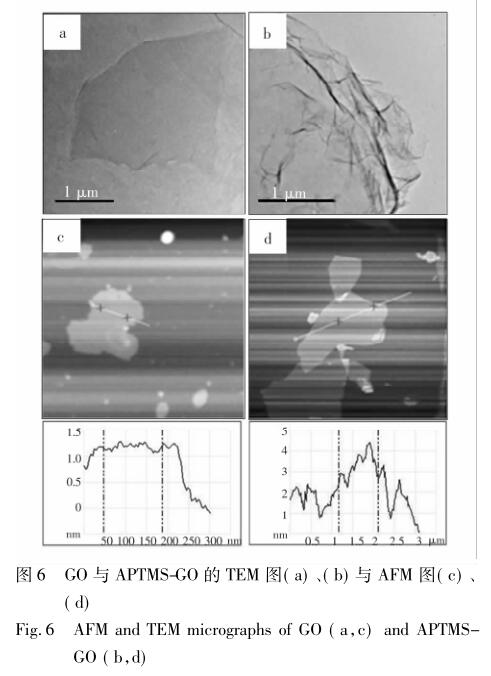
图 6 为 GO 与 APTMS-GO 的 AFM 平面二维图 ( a、b) 与 TEM 图( c、d) 。图 6a 和 6b 分别显示了 GO 改性前后微观形貌的变化,可以看到改性后的石墨烯表面出现了起伏与褶皱,这可能是由于经改 性后的石墨烯片层间距变大,变薄,表面能变高,片 层处于热力学不稳定状态所致。而改性前的氧化石 墨烯只是呈现出一种松散、平整而透明的状态。因 此,二者直观上的差异也再次佐证了 APTMS 与 GO 之间发生了反应。图 6c 和 6d 两图则直观揭示了 GO 和 APTMS-GO 片层的厚度及横向尺寸; 从内置 曲线图上可以看到,改性后的片层厚度由原来 GO 的 1 ~ 2 nm 变化到 APTMS-GO 的 2 ~ 4 nm,说明 APTMS 确实与 GO 发生了反应。以上结果与之前 的 XRD 分析高度吻合。
2. 1. 6 XPS 分析
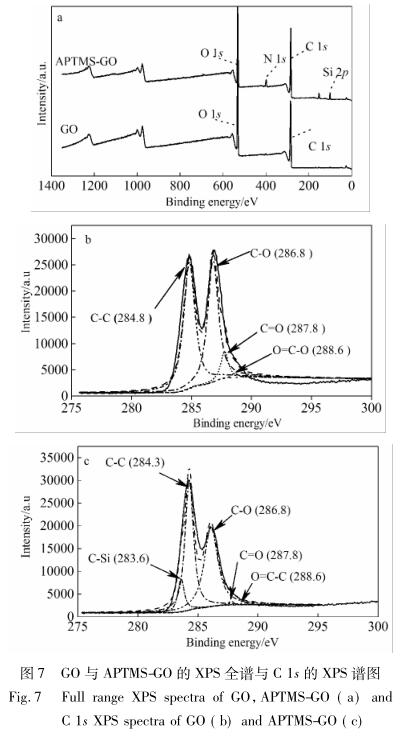
为了考察改性前后石墨烯片层上的价态变化, 对 GO 和 APTMS 进行了 XPS 测试,结果见图 7。
从图 7a 可以看到,相对于 GO 上只有 C 1s 和 O 1s,APTMS-GO 则出现了 N 1s 和 Si 2p,说明 APTMS 被成功地嫁接到了 GO 上。从二者的 C 1s 谱图( 图 7b) 可知,位于 284. 8、286. 8、287. 8 和 288. 6 eV 的 峰 [21] 分别对应于 GO 的 C—C、C—O、 C O 和 O C—C,而 相 比 之 下,图 7c 中 APTMS-GO 在 C—O和 C O 两处出现峰强度的一致降低,同时伴 随着 C—Si( 283. 6 eV) 新峰 [21] 的出现。这些结果再 一次说明,APTMS 与 GO 发生了反应,且与之前的 FTIR、XRD、TG、Raman 的分析保持一致。
2. 2 APTMS-GO/WPU 纳米复合膜的表征
2. 2. 1 FTIR 分析
为了分析聚氨酯改性前后的化学结构,利用全反射红外光谱仪对其进行了表征,结果如图 8 所示。
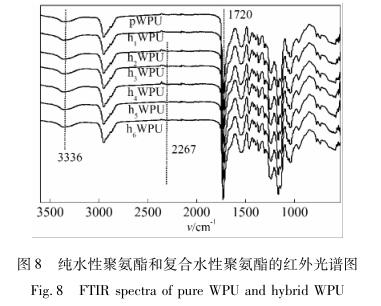
从图 8 可以看出,—NCO 在2 267 cm征吸收峰基本消失,相继出现的是3 336 cm -1 处氨 基甲酸酯基中—NH 的伸缩振动峰,与未改性的聚 氨酯( pWPU) 红外光谱相比,复合产物的红外谱图 并无明显变化,有可能是由于加入的 APTMS-GO 含 量低,且 APTMS-GO 的红外特征峰与聚氨酯的特征 峰发生了重叠。
2. 2. 2 TEM 形貌观察和粒径分析
用 TEM 观察乳液粒子形貌,结果如图 9 所示。
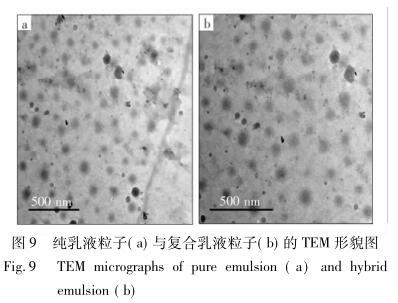
从图 9 可以看出,APTMS-GO 加入后,粒径出现 了细微的变化,尺寸有所增大,可能是氨基化石墨烯 与聚氨酯链段之间新生的氨基甲酸酯键与原有聚氨 酯软段之间产生了额外的氢键作用,在很大程度上 促使了聚合物的交联,从而使粒径有所增大; 另一方 面,WPU 成盐后的—COO - 、NH 4 + 与 APTMS-GO 之 间存在静电斥力与位阻效应,使得在乳化阶段聚合 物自身发生了缠绕,也促使了粒径的增大。 图 10 是纯净 WPU 和不同 APTMS-GO 含量的 复合乳液粒径区间分布图。
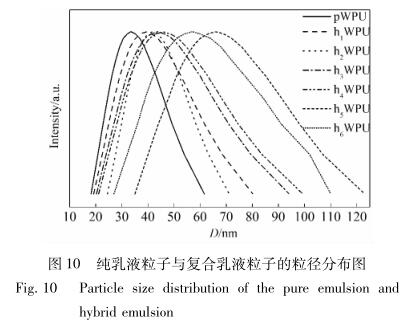
由图 10 可知,纯聚氨酯乳液的平均粒径在 33. 5 nm,且粒径分布较窄,随着 APTMS-GO 含量的 增加,聚氨酯乳液粒径稍有增大的趋势,粒径分布有 所加宽,从 h 1 WPU 乳液的 38. 9 nm 逐渐增大到 h 6 WPU 的 66. 2 nm,原因可能是 APTMS-GO 上的羟 基、羧基与氨基与 WPU 硬段之间新增的氢键破坏 了原有聚合物软硬链段,使层数较少的 APTMS-GO 能够稳定分散于聚合物中,从而使粒径有大有小,平 均粒径尺寸随着 APTMS-GO 添加量的增大而增大; 此外,APTMS-GO 在聚合物中会发生团聚,从而促进 聚合物交联,也会使粒径变大。综上所述,随着 APTMS-GO 含量的增加,聚氨酯乳液粒径有增大的 趋势。
2. 2. 3 热分析 图 11 为纯水性聚氨酯和复合聚氨酯的热失重 曲线。
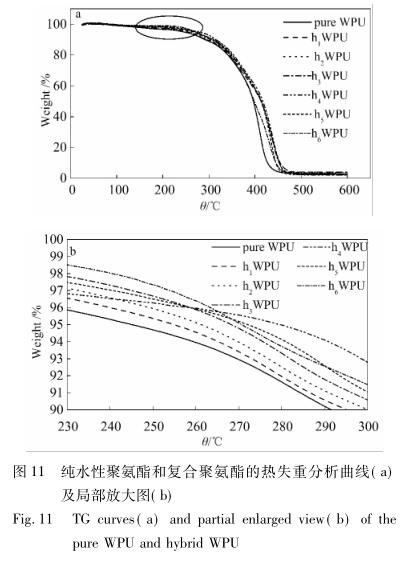
嵌段共聚物 WPU 在 N 2 氛围下的热降解分为 两个阶段: 第一阶段是在 250 ℃ 附近硬段( 主要为 氨基甲酸酯键) 的分解,另一个阶段是在 350 ℃ 附 近软段的分解 [6 -9] 。从图 11( a) 可以观察到,纯水 性聚氨酯与复合水性聚氨酯的热分解趋势相似,但 是 APTMS-GO/WPU 的热分解温度相对于 pWPU 在 热分解第一阶段出现了延迟,随着 APTMS-GO 含量 的逐渐增加这种延迟愈加明显。当 APTMS-GO 质 量分数为 0. 22%时,此现象表现得最为显著。在热 重分析中,采用热失重 5% ( T d5 ) 为初始热降解温 度,从局部放大图 11b 可以看到,在 T d5 时各试样的热降解温度如下: pWPU 为 245 ℃; h 1 WPU 为 254 ℃; h 2 WPU 为 261 ℃; h 3 WPU 为 268 ℃; h 4 WPU 为 279 ℃; h 5 WPU 为 272 ℃; h 6 WPU 为 270 ℃。由此 可见,随着 APTMS-GO 含量的增加,复合聚氨酯的 T d5 先提高后下降,尤其在 APTMS-GO 质量分数为 0. 22%时,T d5 提高了 34 ℃,可能是由于 APTMS-GO 与聚氨酯链段之间产生了脲基甲酸酯键及其衍生出 的氢键,使得无机物 APTMS-GO 能够以共价键的方 式在有机 - 无机界面间相结合,最终均匀地分散到 聚合物基体中。当复合材料热分解时,由于氨基化 石墨烯热绝缘能力被转移到了有机 - 无机两相界 面,促使软硬段间的氨基甲酸酯键断裂需要更多的 热量,最终在一定程度上抑制了聚氨酯乳胶膜的热 分解。但随着 APTMS-GO 含量的继续增加,会造成 聚氨酯软硬段相分离度增大,在一定程度上减少了 氨基甲酸酯键和氢键的数量,最终导致了复合物的 热稳定性变差 [23] 。
2. 2. 4 力学分析
不同 APTMS-GO 含量水性聚氨酯的拉伸曲线
如图 12 所示。
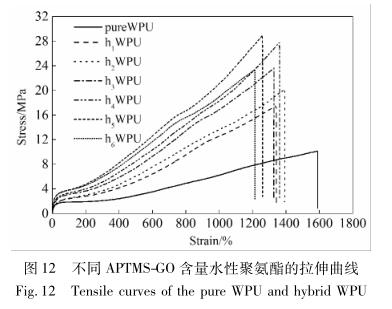
由图 12 可见,随着 WPU 中 APTMS-GO 的不断 加入,拉伸强度均有所增加,当 APTMS-GO 添加量 为 0. 33%时,拉伸强度由 pWPU 的 10. 13 MPa 提高 到了最大值 28. 96 MPa。可能是由于 APTMS-GO 在 聚合物中实现了有效而均匀的分散,APTMS-GO 与 聚氨酯链段之间形成的共价键以及石墨烯本身均具 备大的比表面积,使得 APTMS-GO 与 WPU 主体间 的界面作用力得到大大增强,在拉伸过程中发生了 一定程度上的应力转移; 另外,APTMS-GO 上未发生 反应的含氧官能团( 如羟基、羧基、羰基、环氧基) 与 WPU 链段上的氨基甲酸酯键存在较强的氢键作用。 所以复合材料的拉伸强度有显著地改善 [24] 。相反 地,伴随着 APTMS-GO 添加量的逐步增加,复合材 料的断裂伸长率则出现了下降,在 APTMS-GO 质量分数为 0. 55%时出现了最小值 1210%,可能是因为 石墨烯本身的刚性,在 APTMS-GO 含量增加的同 时,这种刚性表现在复合材料中愈突出,从而使断裂 伸长率有所下降 [25] 。
2. 2. 5 疏水性测试
图13 为不同 APTMS-GO 含量复合膜的接触角。
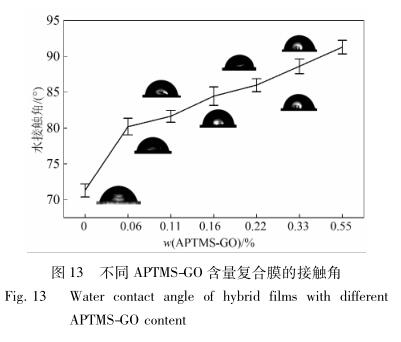
从图 13 可见,加入氨基化石墨烯后,乳胶膜的 接触角明显增大,由纯水性聚氨酯的 71. 3°增大到 APTMS-GO 质量分数为 0. 55% 时的 91. 28°。一方 面,可能是由于在较低浓度的纳米物质掺杂下,纳米 粒子在涂膜表面形成了可以捕捉空气的微纳米结构 层,使滴液不能渗入膜孔,从而提高了膜表面的水接 触角 [26] ; 另一方面,可能是因为 APTMS-GO 与聚氨 酯链段反应后,疏水组分引入到乳胶膜中,使得复合 材料的表面能降低,最终使接触角有所提高。
2. 2. 6 断面观察
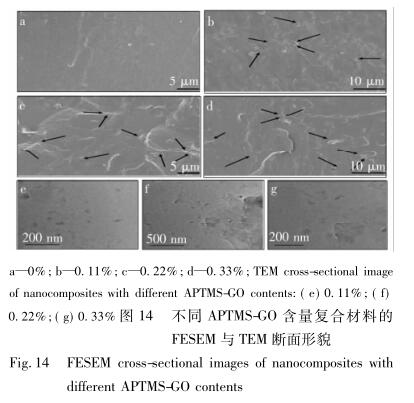
图 14 为不同 APTMS-GO 含量复合材料的断面形貌图。
聚合物的各项性能在很大程度上取决于纳米填 充物质在聚合物中的分散情况,以及二者间的界面 作用力,为此,本文利用 FESEM 来观察复合材料的 断面,来获取 APTMS-GO 在 WPU 主体中的分散情 况。如图 14( a) 所示,纯水性聚氨酯断面平整光滑, 随着 APTMS-GO 的不断填充,断面开始粗糙,掺入 量在 w( APTMS-GO) = 0. 33% 之前,APTMS-GO 能 够在聚氨酯主体中实现较好的分散与嵌入,这可能 是因为在较低 APTMS-GO 含量下,GO 与 WPU 间的 作用力( 主要是少量的共价键和大部分的氢键) 促 使了氨基化石墨烯的有效分散与嵌入。添加量从 0. 10%增到 0. 33%的过程中,发现截面之中的亮色 区域增多,“海 - 岛”结构越来越明显,可能是由于 氨基化石墨烯与聚氨酯链段之间形成氨基甲酸酯键 后,—NH—CO 与软段之间形成了新的氢键,从而破坏了原有聚氨酯软硬段间的作用力,引起了软硬段 的微相分离 [23] ,并发生了团聚。随着 APTMS-GO 含 量的继续加大,团聚会越来越严重,这是因为在 APTMS-GO 含量较大时,由于纳米粒子具备很高的 比表面积和比表面能,存在的引力作用导致粒子间 极易发生团聚现象,图 14( e、f、g) 中也显示出了纳 米物质的分散情况,其分布状况与 FESEM 中的状况 保持一致。
3 结论
利用制备的氨基化石墨烯,以原位聚合的方式 与聚氨酯预聚物反应,成功制备了热稳定性较强、力 学性能好、疏水性优异的 APTMS-GO/WPU 复合物。
复合物的红外以及复合乳液粒子 TEM 表明: APTMS-GO 的添加对聚氨酯主要结构和乳液粒径无 较大影响。涂膜断面的 FESEM 则显示,低含量的 APTMS-GO 能够在 WPU 主体中实现有效的分散,从 而促进了热学、力学性能的改善,但当 APTMS-GO 质量分数超过 0. 33% 后则会发生微相分离和局部 的团聚,从而对聚合物原有的软硬段结构有所影响。
由于氨基化石墨烯的有效分散以及有机 - 无机 相间产生的共价键,石墨烯上的一部分应力和热绝 缘能力被转移到了聚氨酯链段上,其中复合材料的 拉伸强度伴随着断裂伸长率的降低而有所提高。当 APTMS-GO 的质量分数为 0. 33% 时,拉伸强度出现 了最大值 28. 96 MPa,而 TG 结果显示,材料的初始 热降解温度 T d5 从 pWPU 的 245 ℃增大到了 h 4 WPU 的 279 ℃,初始分解温度提高了34 ℃。疏水测试表 明: 由于无机纳米材料的添加,导致复合物表面构成 微纳米结构,从而在一定程度上实现了分子结构中 疏水性的改善。
参考文献:
[1] Engels H W,Pirkl H G,Albers R,et al. Polyurethanes: versatile materials and sustainable problem solvers for today' s challenges [J]. Angewandte Chemie International Edition,2013,52 ( 36) : 9422 -9441.
[2] Wu G,Kong Z,Chen J,et al. Preparation and properties of waterborne polyurethane/epoxy resin composite coating from anionic terpene-based polyol dispersion[J]. Progress in Organic Coatings,2014,77( 2) : 315 -321.
[3] Elrebii M,Mabrouk A B,Boufi S. Synthesis and properties of hybrid alkyd-acrylic dispersions and their use in VOC-free waterborne coatings[J]. Progress in Organic Coatings,2014,77 ( 4) : 757 -764.
[4] Sardon H,Irusta L,Aguirresarobe R H,et al. Polymer/silica nanohybrids by means of tetraethoxysilane sol-gel condensation onto waterborne polyurethane particles[J]. Progress in Organic Coatings,2014,77( 9) : 1436 -1442.
[5] Wu Z,Wang H,Tian X,et al. Surface and mechanical properties of hydrophobic silica contained hybrid films of waterborne polyurethane and fluorinated polymethacrylate[J]. Polymer,2014, 55( 1) : 187 -194.
[6] Zhao C X,Zhang W D,Sun D C. Preparation and mechanical properties of waterborne polyurethane/carbon nanotube composites [J]. Polymer Composites,2009,30( 5) : 649 -654.
[7] Kwon J,Kim H. Comparison of the properties of waterborne polyurethane/multiwalled carbon nanotube and acid-treated multiwalled carbon nanotube composites prepared by in situ polymerization[J]. Journal of Polymer Science Part A: Polymer Chemistry,2005,43( 17) : 3973 -3985.
[8] Zhou H,Wang H,Tian X,et al. Preparation and properties of waterborne polyurethane/antimony doped tin oxide nanocomposite coatings via sol-gel reactions[J]. Polymer Composites,2014,35 ( 6) : 1169 -1175.
[9] Chen S L,Chen S Z,Zhao G H,et al. Fabrication and properties of novel superparamagnetic,well-dispersed waterborne polyurethane/ Ni-Zn ferrite nanocomposites [J]. Composites Science and Technology,2015,119: 108 -114.
[10] Fu H,Yan C,Zhou W,et al. Preparation and characterization of a novel organic montmorillonite/fluorinated waterborne polyurethane nanocomposites: Effect of OMMT and HFBMA[J]. Composites Science and Technology,2013,85: 65 -72.
[11] Gao X,Zhu Y,Zhou S,et al. Preparation and characterization of well-dispersed waterborne polyurethane/CaCO 3 nanocomposites [J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects,2011,377( 1) : 312 -317.
[12] García-Pacios V,Jofre-Reche J A,Costa V,et al. Coatings prepared from waterborne polyurethane dispersions obtained with polycarbonates of 1,6-hexanediol of different molecular weights [J]. Progress in Organic Coatings,2013,76( 10) : 1484 -1493.
[13] Huang T,Xin Y,Li T,et al. Modified graphene/polyimide nanocomposites: reinforcing and tribological effects[J]. ACS Applied Materials & Interfaces,2013,5( 11) : 4878 -4891.
[14] Yang S Y,Lin W N,Huang Y L,et al. Synergetic effects ofgraphene platelets and carbon nanotubes on the mechanical andthermal properties of epoxy composites[J]. Carbon,2011,49(3) : 793 -803.
[15] Seyedin M Z,Razal J M,Innis P C,et al. Achieving outstanding mechanical performance in reinforced elastomeric composite fibers
using large sheets of graphene oxide[J]. Advanced Functional
Materials,2015,25( 1) : 94 -104.
[16] Li J,Ye F,Vaziri S,et al. Efficient inkjet printing of graphene
[J]. Advanced Materials,2013,25( 29) : 3985 -3992.
[17] Balandin A A,Ghosh S,Bao W,et al. Superior thermal
conductivity of single-layer graphene [J]. Nano Letters,2008,8
( 3) : 902 -907.
[18] Scheuermann G M,Rumi L,Steurer P,et al. Palladium
nanoparticles on graphite oxide and its functionalized graphene
derivatives as highly active catalysts for the Suzuki-Miyaura
coupling reaction[J]. Journal of the American Chemical Society,
2009,131( 23) : 8262 -8270
[19] Du J,Lai X,Yang N,et al. Hierarchically ordered macro-
mesoporous TiO 2 -graphene composite films: Improved mass
transfer,reduced charge recombination,and their enhanced
photocatalytic activities[J]. ACS Nano,2010,5( 1) : 590 -596
[20] Matsuo Y,Tabata T,Fukunaga T,et al. Preparation and
characterization of silylated graphite oxide[J]. Carbon,2005,43
( 14) : 2875 -2882.
[21] Matsuo Y,Nishino Y,Fukutsuka T,et al. Introduction of amino
groups into the interlayer space of graphite oxide using 3-
aminopropylethoxysilanes[J]. Carbon,2007,45 ( 7) : 1384 -
1390.
[22] Liao W H,Yang S Y,Hsiao S T,et al. Effect of octa
( aminophenyl ) polyhedral oligomeric silsesquioxane
functionalized graphene oxide on the mechanical and dielectric
properties of polyimide composites[J]. ACS Applied Materials &
Interfaces,2014,6( 18) : 15802 -15812.
[23] Zou J,Zhang F,Huang J,et al. Effects of starch nanocrystals on
structure and properties of waterborne polyurethane-based
composites[J]. Carbohydrate Polymers,2011,85( 4) : 824 -831.
[24] Liang J,Huang Y,Zhang L,et al. Molecular-level dispersion of
graphene into poly ( vinyl alcohol) and effective reinforcement of
their nanocomposites[J]. Advanced Functional Materials,2009,
19( 14) : 2297 -2302.
[25] Wang X,Xing W,Song L,et al. Fabrication and characterization
of graphene-reinforced waterborne polyurethane nanocomposite
coatings by the sol-gel method [J]. Surface and Coatings
Technology,2012,206( 23) : 4778 -4784.
[26] Lakshmi R V,Bharathidasan T,Bera P,et al. Fabrication of
superhydrophobic and oleophobic sol-gel nanocomposite coating
[J]. Surface and Coatings Technology,2012,206( 19) : 3888 -
3894.