两种不同化学组成的超支化聚氨酯改性环氧树脂*
董慧慧 朱新远**(上海交通大学化学化工学院 上海 200240)
摘 要 利用芳香族二元异氰酸酯和脂肪族二元异氰酸酯分别与二乙醇胺反应,设计合成了不同化学组成 的超支化聚氨酯,考察了其对环氧树脂的改性作用. 运用核磁共振、傅里叶红外光谱(FTIR)、扫描电子显微 镜(SEM)、弯曲、拉伸、冲击强度、储能模量、示差扫描量热法(DSC)以及热失重(TGA)等对超支化聚氨酯 的结构及其性能进行了证实和表征,并发现基于芳香族超支化聚氨酯改性的环氧树脂共混体系表现出均相 结构,而脂肪族超支化聚氨酯和环氧树脂共混后却形成了非均相体系. 均相的共混体系证明超支化聚合物自 身能够有效地改善环氧树脂的力学性能和热学性能.
关键词 超支化聚氨酯,环氧树脂,改性,微相结构
环氧树脂是一类重要的高分子材料,具有良 好的粘接性、耐酸碱性和热力学性能,广泛应用 于涂料、黏合剂、复合材料等领域. 然而,环氧 树脂固化后韧性差,这在很大程度上限制了它的 应用范围 [1] . 目前环氧树脂的有效增韧方法主要 包括橡胶粒子增韧、核壳结构聚合物增韧、刚性 高分子原位聚合增韧等,但同时也带来了易老化 降解、工艺性能差等问题 [2~7] . 近年来,人们尝试 采用高度支化聚合物共混增韧环氧树脂. 超支化 聚合物是一类可以工业化大规模合成的高度支 化聚合物,具有独特的三维准球状结构,在共混 体系中通过吸收分散能量起到内增韧效果 [8] ;超 支化聚合物末端带有大量活性基团,可提供良好 的界面相互作用或直接参与固化反应形成交联 网络,这些都为增韧环氧树脂提供了理论基础. 迄今为止,人们已经采用商业化的超支化聚酯 Boltorn™作为增韧剂来改性环氧树脂 [9, 10] ,取得 了良好的增韧效果. 但这一体系呈现微观相分离 状态,存在明显的空穴. 由于微观相分离结构可 以改善环氧树脂的性能 [11] ,因此难以直接说明超 支化聚合物自身在体系中的增韧作用. 此外,微 观相分离尽管改善了共混材料的力学性能,却损 害了环氧树脂的模量和热性能,影响了树脂的实 际应用. 为了尽可能减少对其他性能的影响,人 们提出利用反应型超支化聚合物来增韧环氧树 脂,但其合成复杂,控制困难,成本升高. 与现 有方法不同,本文制备了具有均相结构的芳香族 超支化聚氨酯共混增韧环氧树脂,并以非均相结 构的脂肪族超支化聚氨酯共混增韧体系作为对 照,直接证明超支化聚合物自身可以改善环氧树 脂力学性能,同时提高了环氧树脂固化物的模量 和热性能.
1 实验部分
1.1 试剂 六亚甲基二异氰酸酯(HDI),分析纯,上海 阿达玛斯试剂;4,4-二苯基甲烷二异氰酸酯 (MDI),分析纯,Acros试剂;二乙醇胺(DEOA), 分析纯,阿拉丁试剂;N,N-二甲基甲酰胺(DMF)、 N,N- 二 甲 基 乙 酰 胺 (DMAc) 、 二 甲 基 亚 砜 (DMSO),分析纯,上海试剂公司,使用前减压 蒸馏;无水乙醚、三乙烯四胺,分析纯,上海试 剂公司;环氧树脂E44,上海元邦化工制造有限 公司.
1.2 分析测试方法
核磁谱图(NMR)测试采用Varian MERCURY plus 400核磁共振仪,DMSO-d 6 作为氘代溶剂.
红外光谱(FTIR)采用美国Perkin Elmer公司 的PE-1000型红外光谱仪,扫描范围为4000 ~ 500 cm −1 ,将样品与KBr研磨均匀后压片制样.
采用美国美特斯公司的Criterion 43型电子万 能试验机进行拉伸、三点弯曲测试,拉伸夹具间 距为40 mm,试验速度为5 mm/min,弯曲跨度为 64 mm,试验速度为10 mm/min. 采用英国 RAY-RAN公司的摆锤式冲击试验机进行无切口 悬臂梁式冲击测试,落锤的质量为0.818 kg,下落 速度为3.5 m/s.
动态热机械分析(DMA)采用美国Perkin Elmer公司的DMA 8000的动态热分析仪,测试方 法为单悬臂振动法,样品尺寸为25 mm × 10 mm × 1.7 mm,测试温度范围为0 ~ 140 °C,升温速率 3 K/min,应变振幅为0.05%,频率为1 Hz.
示差扫描量热分析(DSC)采用美国TA公司的 Q2000型热分析仪,升温速率为20 K/min,N 2 氛 围下进行.
热 失 重 分 析 (TGA) 采 用 美 国 TA 公 司 的 Q5000IR型热分析仪,测试温度范围为50 ~ 800 °C,升温速率为20 K/min,N 2 氛围下进行.
采用英国马尔文公司的Viscotek TDA 305型 凝胶渗透色谱仪(GPC)测定聚合物分子量及分子 量分布,用加入LiBr的DMF做洗脱剂,淋洗速度 为1 mL/min,测试温度为25 °C.
采用美国FEI公司的Nova Nano SEM 450型 扫描电子显微镜(SEM)拍摄扫描电镜图,样品表 面进行喷金处理.
1.3 超支化聚氨酯的合成
1.3.1 芳香族超支化聚氨酯(M-HBPU)的合成
将1.05 g (10 mmol) DEOA加入到圆底烧瓶 中,加入5 mL DMSO和5 mL DMAc溶解. 称取 2.5 g (10 mmol) MDI,用5 mL DMAc溶解后在N 2 保护下滴加到烧瓶中. 0 °C下先滴加3 mL溶液反 应1 h,滴加剩余溶液继续反应4 h,升温至60 °C 反应6 h. 反应结束后,用500 mL无水乙醚沉淀, 再用少量DMF溶解,乙醚沉淀,重复操作多次后 真空干燥 [12] .
1.3.2 脂肪族超支化聚氨酯(H-HBPU)的合成
将1.05 g (10 mmol) DEOA加入到圆底烧瓶 中,加入10 mL DMF溶解. 称取2.5 g (10 mmol) HDI,用10 mL DMF溶解后在N 2 保护下滴加到烧 瓶中. 常温下先滴加3 mL溶液反应1 h,滴加剩余 溶液继续反应4 h,升温至60 °C反应2 h,最后升 温至70 °C反应6 h. 反应结束后,用500 mL无水 乙醚沉淀,洗涤多次后真空干燥 [13] .
1.4 测试样条制备
用少量DMF溶解超支化聚氨酯,溶液与固化 剂三乙烯四胺混合后加入到已脱气泡的环氧树 脂E44中,慢慢搅拌,然后将混合物浇注到模具 中. 以加入的环氧树脂质量为100份计算,超支化 聚氨酯的加入质量份数分别为0、0.5、1、1.5、2, 固化剂三乙烯四胺的质量为11份,40 °C下固化 9 h,80 °C真空条件下放置2 h去除树脂中的 DMF,脱模后进行性能测试,样条尺寸为10 mm × 80 mm × 2 mm. 样条制备过程如图1所示.
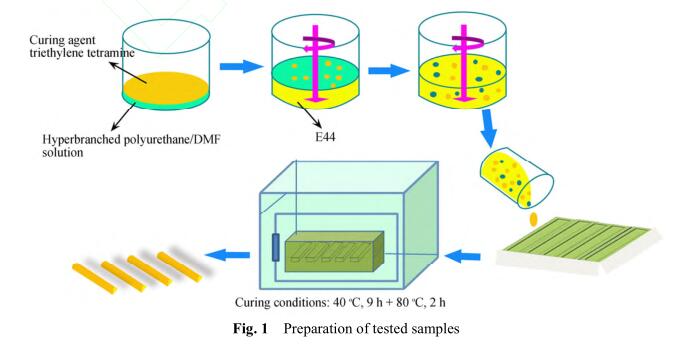
2 结果与讨论
2.1 超支化聚氨酯的合成与结构表征
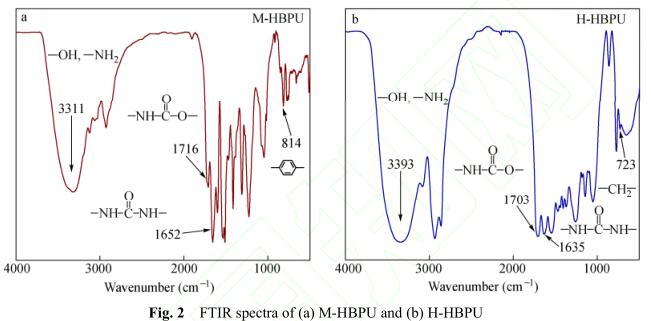
根据文献,可以利用A 2 + CB 2 方法方便地制 备超支化聚氨酯 [12, 13] . 本文采用该方法分别合成 了M-HBPU和H-HBPU,其分子量及分子量分布 参见表1. 产物的红外光谱图如图2所示. 图2(a) 为M-HBPU的吸收谱图,3311 cm −1 处为羟基和氨 基的特征吸收峰,1716 cm −1 为氨基甲酸酯中羰基 的特征吸收峰,1652 cm −1 为脲基中羰基的特征吸 收峰,814 cm −1 为对位取代苯环的特征吸收峰. 图2(b)为H-HBPU的吸收谱图,3393 cm −1 为羟基 和氨基的特征吸收峰,1703 cm −1 为氨基甲酸酯中 羰基的特征吸收峰,1635 cm −1 为脲基中羰基的特 征吸收峰, 723 cm −1 为多个亚甲基相连时的特征 吸收峰. 红外光谱实验结果表明,我们成功地合 成了2类不同分子结构的超支化聚氨酯.

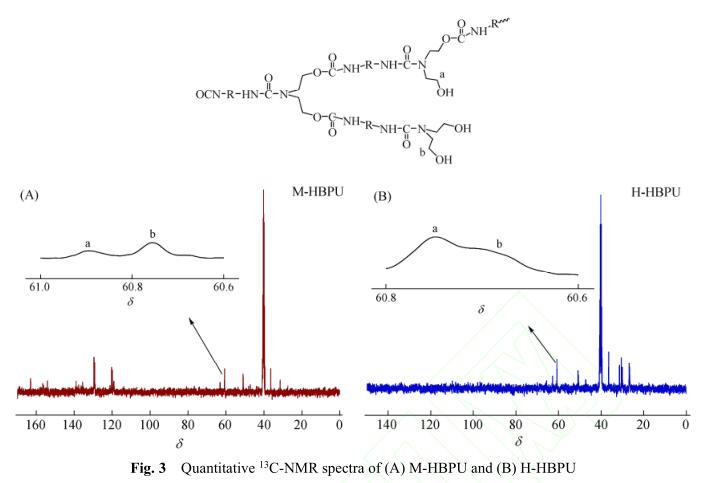
M-HBPU和H-HBPU的定量 13 C-NMR谱图如 图3所示. 基于该定量碳谱,我们可以计算超支化 聚氨酯的支化度(DB). 超支化聚合物中包含3种 结构单元,分别是树形单元(D)、末端单元(T)和 线形单元(L),支化度为树形单元和末端单元在分 子结构单元中所占的比例,是表征超支化聚合物 拓扑结构的一个重要参数. 在本文中,采用A 2 + CB 2 法合成超支化聚氨酯,首先形成AB 2 型中间 体,再通过中间体自缩聚得到聚合产物,所以其 支化度可用如下公式进行计算 [12] :
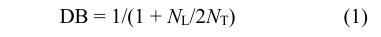
即支化度可只用线形单元数量(N L )和末端单元数 量(N T )的相对量计算. 由于产物的线形单元上包 含1个羟基,末端单元上包含2个羟基,所以 13 C-NMR谱图上线形单元和末端单元中与羟 基相连的C的化学环境不同,在碳谱上的化学位 移也不同. 通过对相应峰面积进行积分并代入 公式(1)中,可计算超支化聚氨酯的支化度,结果 见表1. 所制备的M-HBPU和H-HBPU的支化度分 别是0.66和0.43,表明我们的确得到了具有高度 支化结构的超支化聚氨酯.
根据DSC实验结果,M-HBPU的玻璃化转变 温度(T g )为83 °C,而H-HBPU的T g 为−42 °C. 这种 T g 上 的 差 异 反 映 了 二 者 分 子 结 构 的 不 同 , M-HBPU中含有大量的刚性苯环结构,限制了分 子链的运动;而H-HBPU中含有柔顺性好的脂肪 链,所以在较低温度下即可发生玻璃化转变.
2.2 改性环氧树脂的弯曲断面及微相结构
通过比较纯环氧树脂和改性环氧树脂弯曲 断面的粗糙程度可初步判断材料的韧性;通过分 析断面的微相结构可判断共混体系的相容性. 图 4(A)为纯环氧树脂的弯曲断面,图4(B)为加入 0.5%的M-HBPU改性后的环氧树脂弯曲断面,图 4(C)为含0.5%的H-HBPU的环氧树脂弯曲断面, 图4(a)、4(b)、4(c)分别为对应的高倍扫描电镜图, 可观察材料的微观相结构.

从图4(A) ~ 4(C)可以看出,纯环氧树脂的弯 曲断面光滑,为脆性断裂,说明材料的韧性较差; 改性后的环氧树脂断面粗糙,为韧性断裂,且断 面上呈现细小的纤维状,材料的韧性增强,表明 超支化聚氨酯提高了环氧树脂的韧性. 从高倍扫 描电镜图4(a) ~ 4(c)中可以看出,加入M-HBPU 后,环氧树脂为均相体系,不发生相分离现象; 而加入H-HBPU后出现相分离结构,断面结构与 传统的超支化聚合物增韧环氧树脂体系相似,存 在明显的粒子空穴. 这是因为M-HBPU中含有大 量苯环,与E44的结构具有相似性,从而提高了 体系的相容性,共混后得到均相结构.
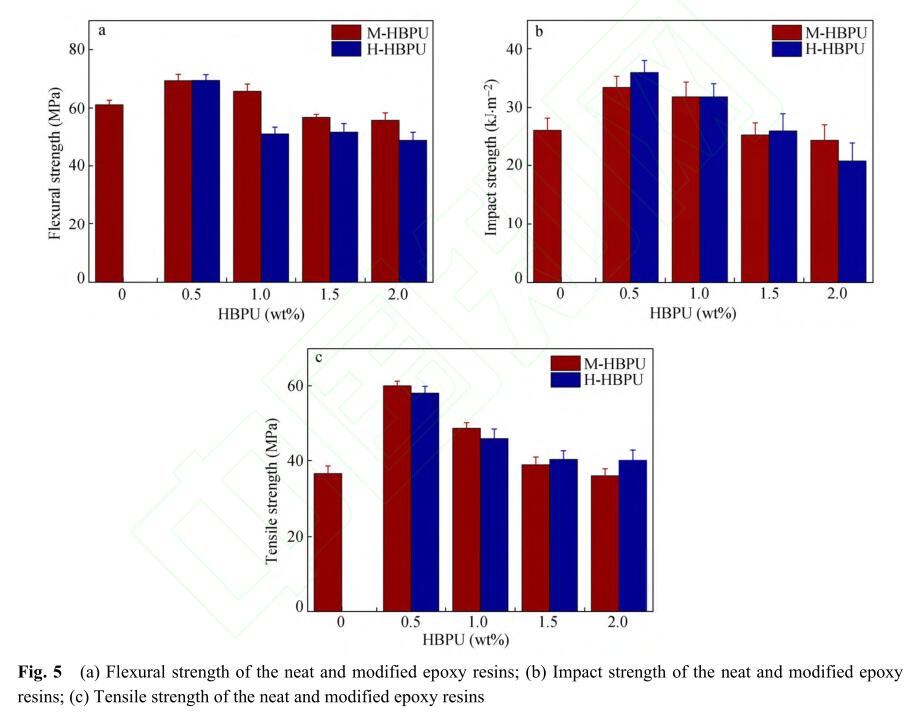
2.3 改性环氧树脂的力学性能
为了探究超支化聚氨酯对环氧树脂固化体 系力学性能的影响,分别对树脂的弯曲韧性和拉 伸强度进行了测试. 不同质量分数的超支化聚氨 酯对环氧树脂韧性的影响如图5所示. 与纯环氧 树脂相比,加入的M-HBPU质量分数为0.5%时, 材料韧性提高得最明显,弯曲强度提高了15.2%, 冲击强度提高了28.8%;H-HBPU加入量为0.5%时,弯曲强度提高了15.6%,冲击强度提高了 38.7%. 从图5(a)和5(b)中可以看出,添加适当的 超支化聚氨酯可以提高固化树脂的韧性,但随着 添加量的增加,材料韧性呈现下降趋势. 当添加 量较低时,超支化聚氨酯通过吸收分散外力达到 增韧效果;当含量增加时,分子链间的链缠结减 少,反而降低了体系的强度. 对比H-HBPU,加 入相同量M-HBPU时树脂的韧性更好,这是因为 加入H-HBPU后体系呈现微相分离,添加量过多 时导致分散相分散不均匀,从而影响连续相的性 能,导致韧性变差. 图5(c)的结果显示,超支化聚 氨酯对环氧树脂的拉伸强度也有影响. 根据表2 可知,加入0.5%的M-HBPU时,拉伸强度从36.62 MPa提高到60.11 MPa,提高了64.15%;加入0.5% 的H-HBPU后,拉伸强度从36.62 MPa提高到58.17 MPa,提高了58.85%. 从实验结果看,加入适量 的超支化聚氨酯可同时提高环氧树脂的韧性和 拉伸强度,这归因于超支化聚合物三维结构可吸 收分散能量. 一旦添加过量超支化聚氨酯,由于 体系的链缠结减少和相分散增多,导致力学性能 下降. 因此存在较佳添加量,可以达到较好的增 强增韧效果.
DMA测试中的储能模量-温度曲线亦可反映 环氧树脂的力学性能 [14] . 如图6所示,温度较低时 可明显看到无论添加哪种超支化聚氨酯,材料的 储能模量(E′)均比纯环氧树脂E′高,这主要是因 为低温下材料处于玻璃态,超支化聚氨酯的立体 椭球状结构可以吸收能量;同时,由于大量的末 端基团可以和环氧树脂形成强烈的次级相互作 用,限制了聚合物链段的运动,从而达到提高存 储能力的效果. 随着温度的升高,链段的运动能 力提升,添加超支化聚氨酯致使链缠结减少、自 由体积增加,这些因素都有利于链段的运动. 因 此,当温度逐渐升高,材料达到玻璃化转变状态 后,同一温度下改性环氧树脂比纯环氧树脂的E′ 低. 然而当超支化聚氨酯质量分数为0.5%时,改 性环氧树脂的E′始终高于纯环氧树脂,说明该组 分对保持甚至提高材料模量的效果较佳.
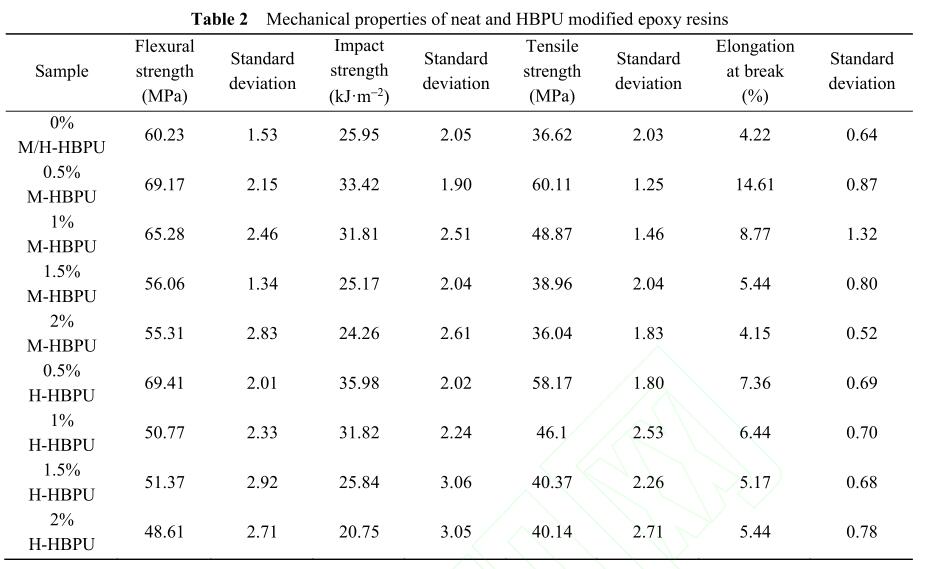
另外,对比图6(a)和6(b)可发现,在低温区, 材料的E′随着加入M-HBPU的质量增加而增加,呈现明显的规律. 而H-HBPU的加入虽然会提高 体系的E′,但因为体系为相分离结构,其模量高 低还与分散相的密度以及分布均匀性有关,所以 E′并不随添加剂含量变化呈现明显的规律性.
2.4 改性环氧树脂的热性能
耐热性能优良是环氧树脂的优势之一,然而 在增韧改性的过程中往往会损失体系的热性能. 例如,采用橡胶粒子改性可以提高环氧树脂的韧 性,但玻璃化转变温度(T g )却大大下降 [15] ;采用 超支化聚合物作为相分离增韧剂,也会出现耐热 性变差 [16] . 为了解决这一问题,人们采用反应型 超支化聚合物来增韧环氧树脂 [17] . 然而与共混方 法相比,设计这样的反应型大分子需对超支化聚 合物进行改性,合成步骤复杂,控制困难,成本 增加. 因此,我们设计合成了含芳香结构的超支 化聚氨酯共混增韧环氧树脂体系(M-HBPU/E44). 一方面,超支化聚合物与双酚A型环氧树脂E44 中都含有大量苯环,结构具有相似性,有助于提 高体系的相容性,从而减少热性能的损失;另一 方面,M-HBPU自身的T g 较高,与纯环氧树脂的 T g 相差不大,更有利于二者相容,从而保持甚至 提高体系的热稳定性. 与此同时,我们也设计了 H-HBPU共混体系(H-HBPU/E44),2个体系的 DSC曲线如图7所示.
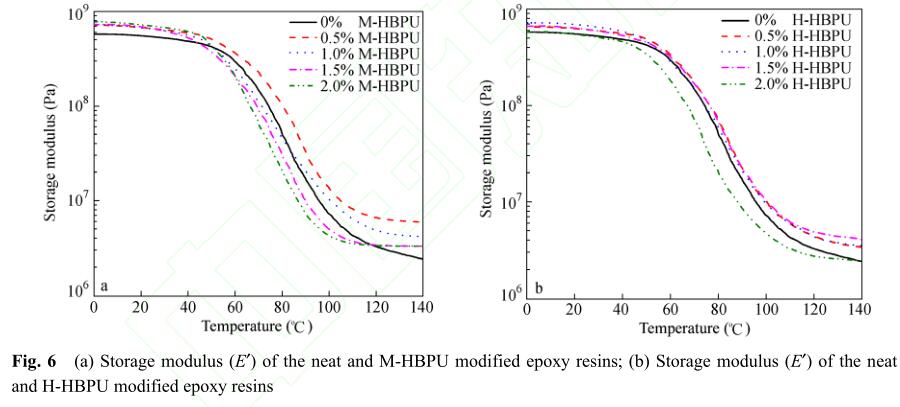
从图7(a)中可以看出,当添加M-HBPU时, M-HBPU/E44体系T g 较纯环氧树脂有所提高. 添 加的质量分数为0.5%时T g 提升最多,环氧树脂的 耐热性得到了提高. 随着超支化聚合物含量增 加,T g 开始下降. 当质量分数达到2%时,其T g 低 于纯环氧树脂. 这是因为超支化聚合物在空间上 呈现三位立体结构,链缠结少 [18] ,引入后会增加 分子链间的自由体积,这些因素都有利于链运 动,因此加入过多超支化聚合物会降低固化物的 热性能. 图7(b)中的DSC曲线为添加H-HBPU后 固化物的玻璃化转变曲线,从图中可以看出,随 增韧聚合物含量增加,T g 逐渐降低,玻璃化转变 范围变宽. 当聚合物的质量分数为2%时,出现了 2个玻璃化转变,说明固化后H-HBPU与环氧树脂 相容性不好,出现微相分离.
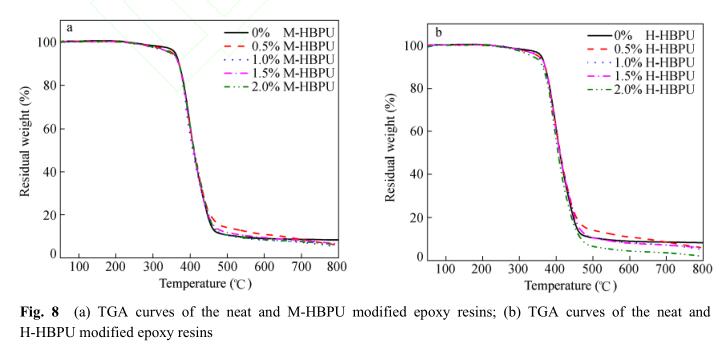
图8给出了M-HBPU/E44和H-HBPU/E44体 系固化后树脂的TGA曲线. 图8(a)为加入了 M-HBPU后固化物的热失重曲线,图8(b)为加入 了H-HBPU后固化物的热失重曲线. 从图中可以 看出,掺杂聚合物后树脂的热失重曲线与纯环氧 树脂的曲线基本重合,这表明体系的耐热性变化 不大. 根据表3中的数据可知,体系热失重10%温 度随聚合物添加量的增加而降低,这主要是因为 超支化聚氨酯的末端含有大量的羟基官能团,随 着温度的升高,羟基间脱水导致失重量增加 [19] , 损失相同重量时的温度下降. 除此之外,失重速 率最大时的温度变化不大,表明加入超支化聚氨 酯后体系的高温热稳定性没有下降.
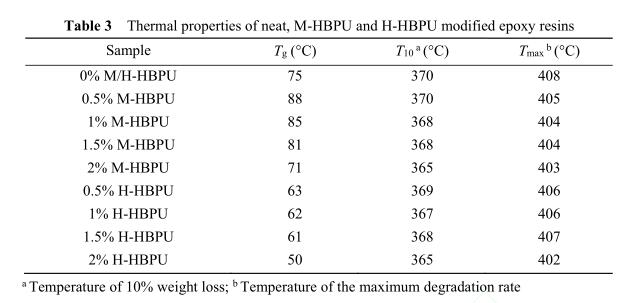
3 结论
我们设计了不同分子结构的超支化聚氨酯用 于环氧树脂的共混改性,探究了超支化聚合物分 子结构对体系力学性能、热性能和微观相结构的 影响. 通过相容性良好的M-HBPU/E44共混体系, 证明利用超支化聚合物自身独特的结构和性能特 点,可以达到增强增韧环氧树脂的效果. 从力学性 能测试的结果可以看出,无论是M-HBPU/E44共 混体系还是H-HBPU/E44共混体系,当超支化聚氨 酯的质量分数为0.5%时,其性能达到较佳. 由于 芳香族聚氨酯与环氧树脂有相似的分子结构和相 近的T g ,提高了共混体系的相容性,维持甚至提 高了材料的热稳定性. 在今后工作中,我们可以通 过设计分子结构,增进超支化聚合物与环氧树脂 的相容性,进一步提高材料的力学性能、热性能 和其他性能.
REFERENCES
1 LeMay J, Kelley F. Adv Polym Sci. Springer: Berlin/Heidelberg, 1986.115 − 1482 Liang Y L, Pearson R A. Polymer, 2010, 51: 4880 − 4890
3 Gan W, Zhan G, Wang M, Yu Y, Xu Y, Li S. Colloid Polym Sci, 2007, 285: 1727 − 1731
4 Chikhi N, Fellahi S, Bakar M. Eur Polym J, 2002, 38: 251 − 264
5 Nguyen F N, Berg J C. Compos Part A: Appl Sci Manufac, 2008, 39: 1007 − 1011
6 Wu S. Polymer, 1985, 26: 1855 − 1863
7 Wilkinson S P, Ward T C, Mcgrath J E. Polymer, 1993, 34: 870 − 884
8 Dodiuk H, Gold Z, Kenig S. J Adhes Sci Technol, 2004, 18: 301 − 311
9 Ratna D, Varley R, Simon G P. J Appl Polym Sci, 2003, 89: 2339 − 2345
10 Foix D, Serra A, Amparore L, Sangermano, M. Polymer, 2012, 53: 3084 − 3088
11 Dou H F, Tian B, Huang Y Z J, Quan Y W, Chen Q M, Yin G. J Adhes Sci Technol, 2016, 30: 642 − 652
12 Gao C, Yan D. Macromolecules, 2003, 36: 613 − 620
13 Zhang Meijie(张美杰), Zhao Xiuli(赵秀丽). Modern Chemical Industry(现代化工), 2013, (9): 56 − 60
14 Jin Q, Misasi J M, Wiggins J S, Morgan S E. Polymer, 2015, 73: 174 − 182
15 Pillai J P, Pionteck J, Häßbler R, Sinturel C, Mathew V S, Thomas S. Ind Eng Chem Res, 2012, 51: 2586 − 2595
16 Luo Kai(罗凯), Su Lin(苏琳), Liu Junhua(刘俊华), Wang Yuechuan(王跃川). Thermosetting Resin(热固性树脂), 2005,
20: 5 − 8
17 Tang B, Liu X B, Zhao X L, Zhang J H. J Appl Polym Sci, 2014, 131: 1107 − 1117
18 Mezzenga R, Boogh L, Månson J A E. Compos Sci Technol, 2001, 61: 787 − 795
19 Jiang Shibao(蒋世宝), Zhang Daohong(张道洪), Jia Demin(贾德民). Adhesion(粘接), 2007, 28: 4 − 6